Single-cell datasets are growing in size and complexity, but existing analytical methods struggle to interpret them at scale.
In raw form, each cell is represented as a high-dimensional matrix of gene expression values, not as structured sequences that language models can interpret.
LLMs cannot work with raw gene-expression matrices directly – they require structured language transformation.
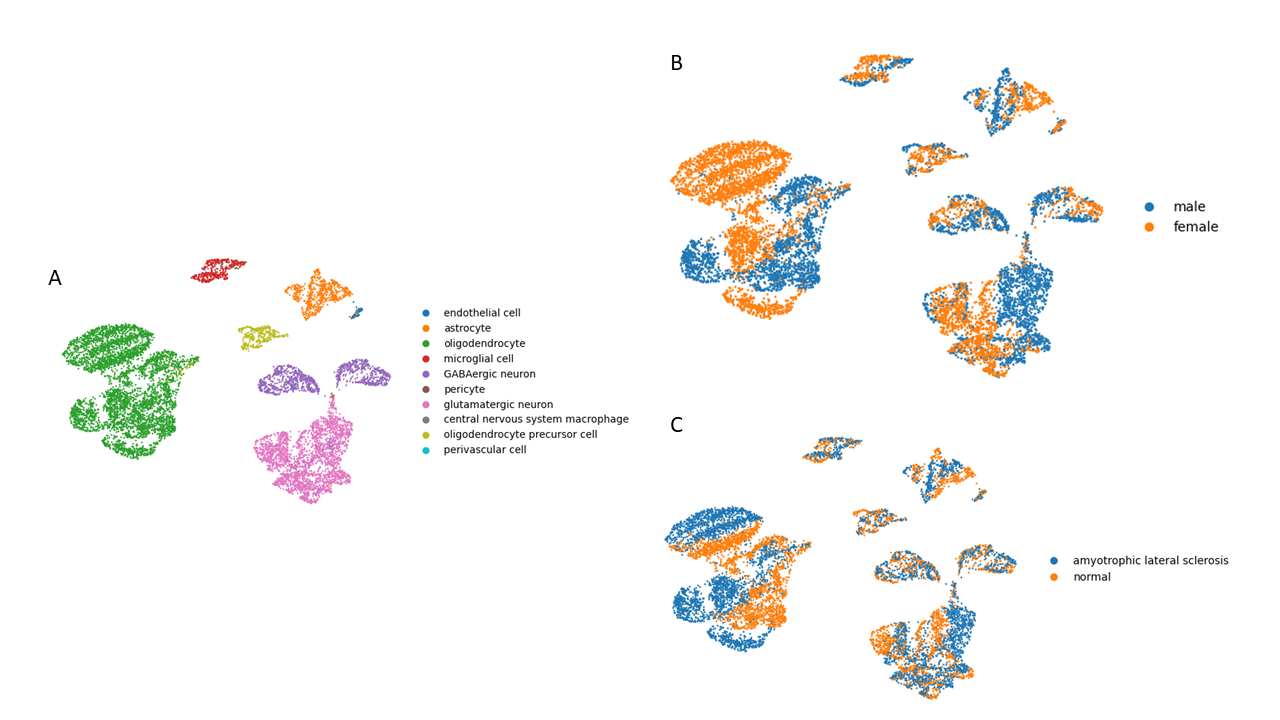
We conducted a structured evaluation of the C2S pipeline on a 10,000-cell ALS motor cortex dataset, measuring reconstruction loss and assessing Gemma-based model performance in supervised cell-type classification.
Raw Single-Cell Data Is Structurally Incompatible with LLMs
Single-cell transcriptomic data is not inherently compatible with language models because it is encoded as high-dimensional numerical matrices rather than structured token sequences.
In its native form, single-cell data is represented as transcript count matrices, where each cell is described by tens of thousands of gene expression values.
In our experimental dataset, after preprocessing and filtering, each of the 10,000 sampled cells was represented across 29,865 genes (originally 35,467 before filtering).
This creates three structural incompatibilities with LLM architectures:
- Continuity and scale – Expression values are continuous numerical quantities, not discrete linguistic tokens.
- High dimensionality – Each cell is defined across tens of thousands of variables, far exceeding standard token windows.
- Non-linear biological structure – Even normalized features often lack linear relationships, complicating both classical ML and deep learning interpretation.
Existing single-cell LLMs (scBERT, GeneCompass, scGPT, scFoundation) attempt to model such data directly but still struggle with:
- Batch effects
- Dropout artifacts
- Sparse matrices
- Rare cell populations
- Zero-shot generalization
Crucially, discretizing continuous gene expression into tokens introduces quantitative precision loss, which affects the ability to distinguish subtle cell states.
A practical example of structural limitation appears even before applying LLMs.
Manual gating strategies in immunology fail to identify rare subpopulations lacking distinct surface markers.
Similarly, standard ML pipelines struggle when relationships between normalized features are non-linear and context-dependent.
In our dataset:
- 75,583 total cells were available
- 10,000 cells were sampled due to infrastructure limits.
- After normalization and filtering, dimensionality remained extremely high (29,865 genes per cell).
This dimensionality illustrates the core incompatibility: LLMs are optimized for sequential token prediction.
The primary limitation in applying LLMs to single-cell biology is representation format.
Until numerical expression matrices are transformed into structured linguistic sequences, LLM architectures remain structurally misaligned with transcriptomic data.
C2S Pipeline and Model Evaluation
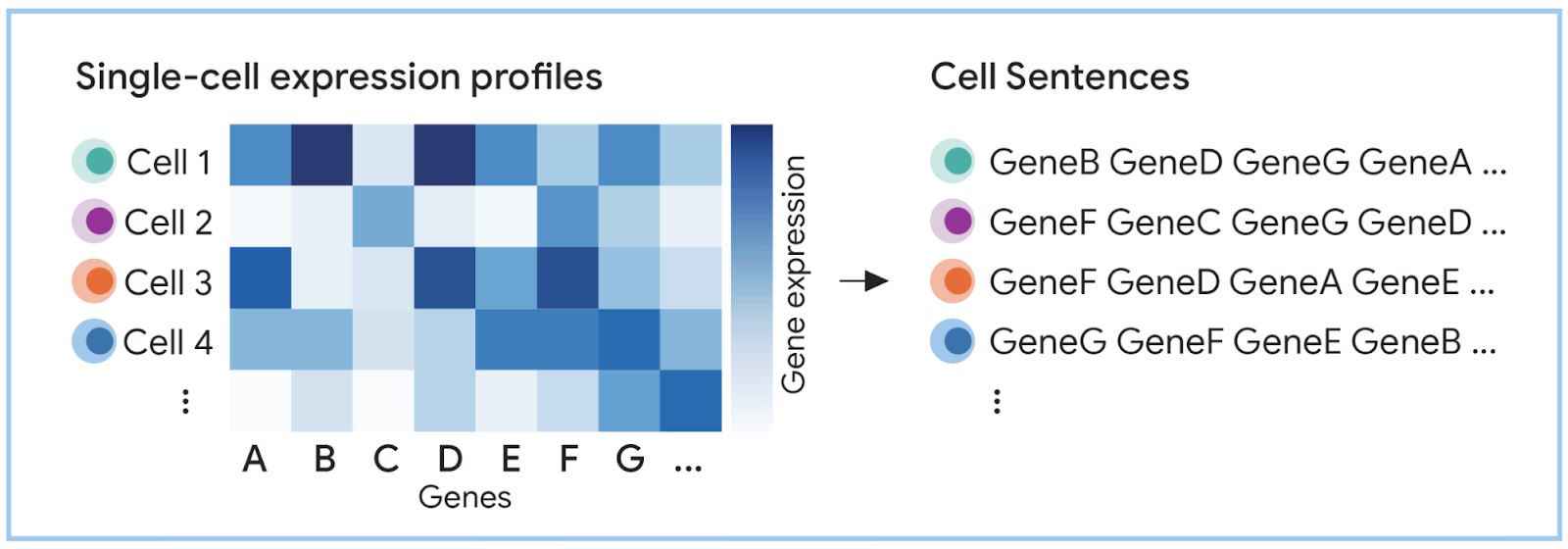
Transforming gene expression profiles into ranked gene “sentences” enables compatibility with language models. However, this transformation does not eliminate biological complexity and introduces measurable trade-offs that depend on model capacity and infrastructure constraints.
The C2S framework converts normalized transcript count matrices into ordered token sequences by ranking genes according to expression level and concatenating gene names into structured “cell sentences”.
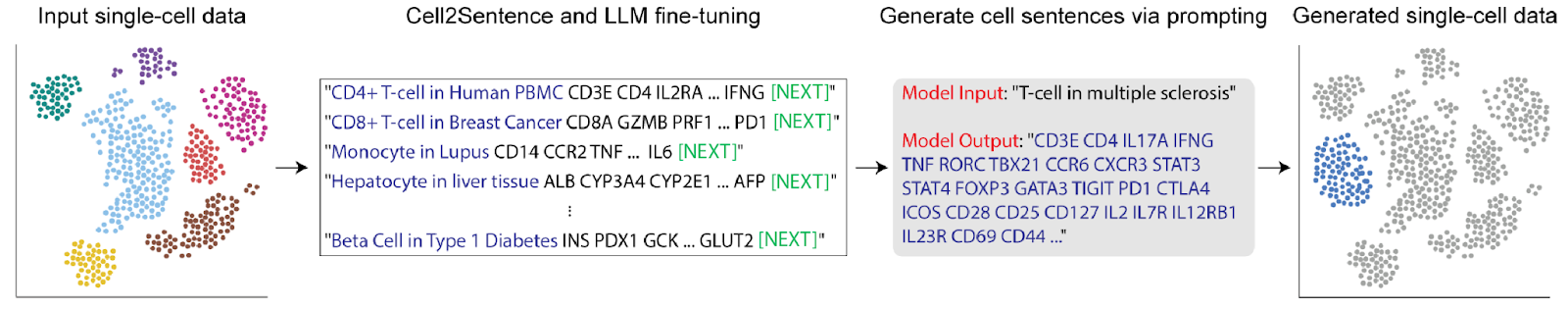
The pipeline implemented in our evaluation consisted of:
- Dataset preprocessing and Scanpy normalization
- Log transformation and rare gene filtering
- Rank-based gene ordering
- Conversion to CSData format
- Reconstruction quality assessment
- Downstream cell-type prediction using Gemma-based models
After subsampling, 10,000 cells were analyzed from an original dataset of 75,583 primary motor cortex cells (ALS and controls).
Following filtering, each cell remained represented across 29,865 genes.
To evaluate information preservation, we reconstructed expression profiles from generated cell sentences using calculated slope (-0.00684) and intercept (0.084275) parameters.
Reconstruction quality assessment indicated that approximately 80% of expression information was preserved after transformation.
However, two constraints remained:
- Reconstruction is not lossless – ranking removes absolute quantitative relationships.
- Model capacity matters – smaller models show reduced ability to detect atypical or rare cell populations.
Due to hardware limitations (Gemma-27B requires ~50GB RAM), evaluation was conducted using C2S-Scale-Gemma-2B.
Using the 10,000-cell ALS motor cortex subset:
- Data retention after C2S conversion ≈ 80%
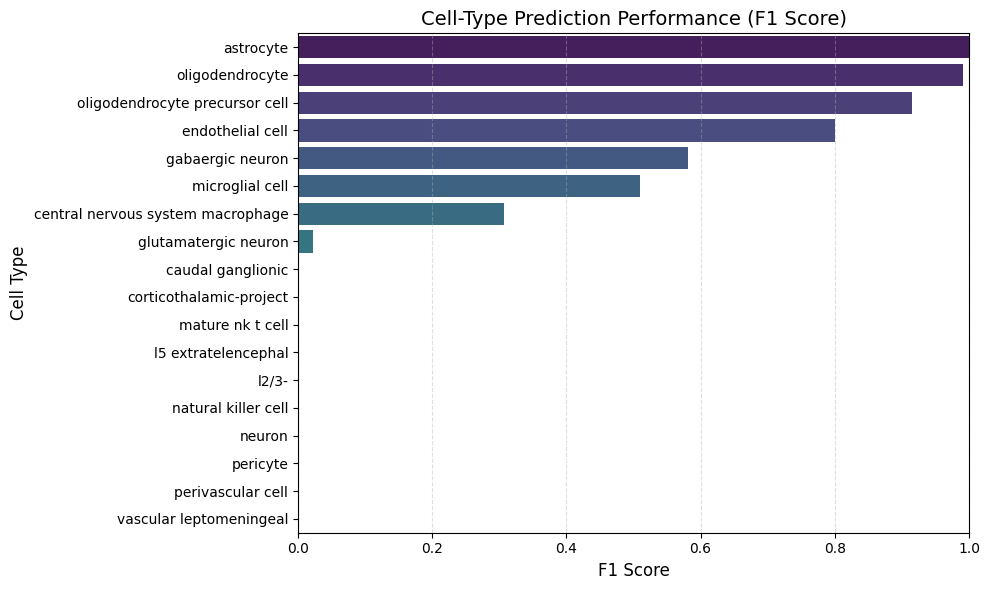
- Common cell types (oligodendrocytes, glutamatergic neurons, astrocytes) achieved F1 scores > 0.5
- Rare or atypical brain cell types showed F1 < 0.5
UMAP comparison between original and reconstructed datasets demonstrated preserved global cluster structure with minor distributional drift.
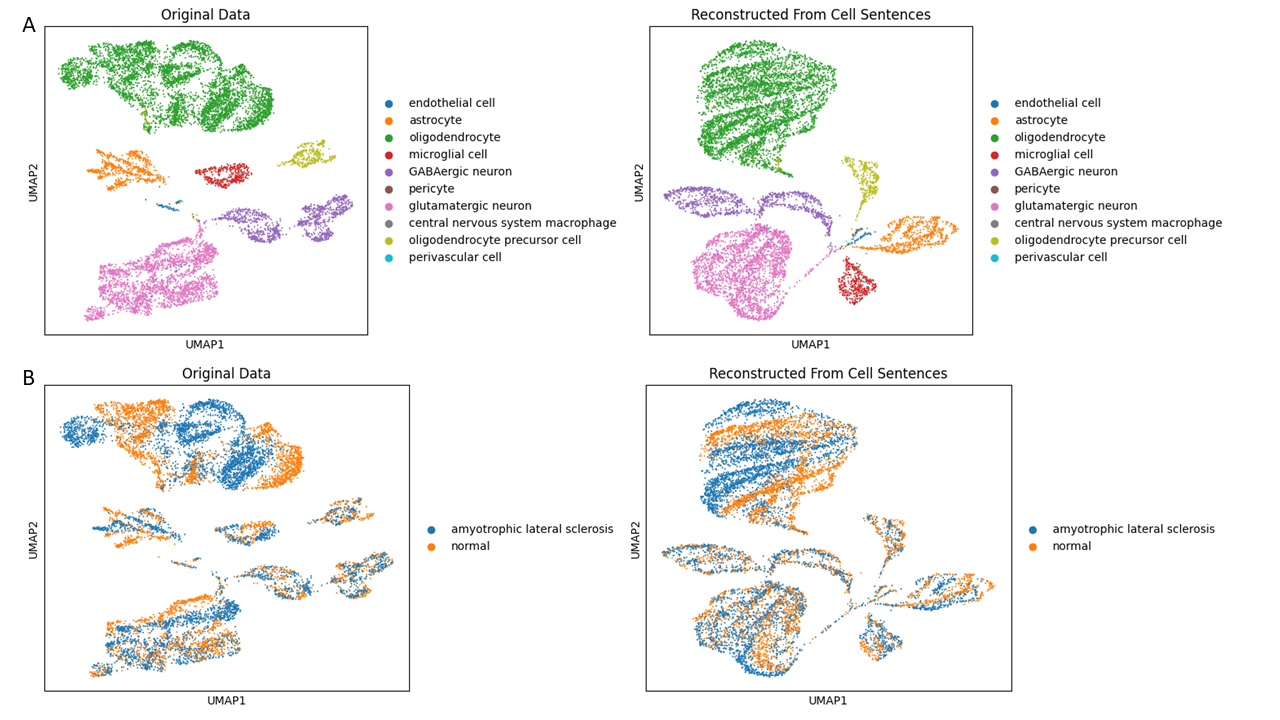
We truncated each cell sentence to the top 300 expressed genes to reduce computational load while keeping the dominant signal.
After C2S conversion:
- ~80% of expression structure was retained
- Global UMAP clusters remained stable
- Minor structural drift was observed
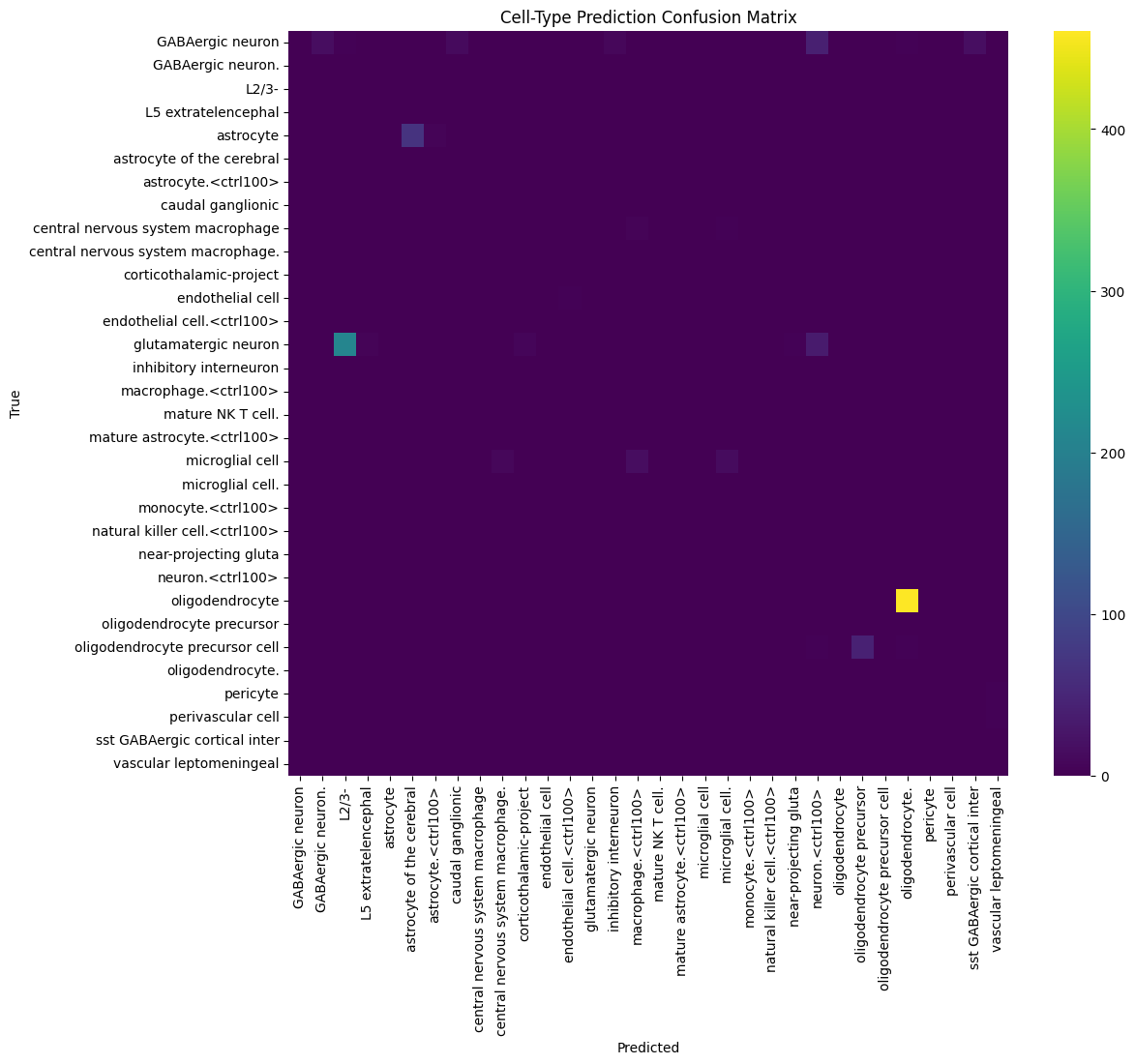
Classification performance showed a clear pattern:
- Major cell types (oligodendrocytes, astrocytes, glutamatergic neurons) → F1 > 0.5
- Mid-frequency populations → moderate performance
- Rare cell types → F1 < 0.5
Errors were concentrated in low-frequency or biologically similar cell populations.
Evaluation was performed using C2S-Scale-Gemma-2B due to infrastructure limits (Gemma-27B requires ~50GB RAM).
C2S makes single-cell data compatible with LLMs while preserving overall biological structure.
Trade-offs remain:
- Ranking reduces quantitative precision
- Smaller models struggle with rare populations
- Performance depends on available compute
Dominant transcriptional programs are preserved. Subtle states are more sensitive to compression.
C2S enables practical LLM-based analysis of single-cell data.
On a 10,000-cell ALS motor cortex dataset:
- ~80% structural retention
- Reliable classification of major cell types
- Rare populations remain challenging under 2B constraints
The main limitation is model capacity. Under realistic hardware settings, C2S delivers biologically meaningful results. Larger models may improve rare-cell detection but significantly increase computational cost.
Conclusion
The C2S framework addresses a fundamental bottleneck in applying large language models to transcriptomic data: representation format.
Our evaluation confirms that:
- Rank-based transformation preserves the majority of global expression structure (~80% retention).
- LLM-based classification of dominant cell types is feasible under constrained infrastructure.
- Performance on rare populations remains sensitive to model capacity and compression effects.
- Scaling to larger models improves potential accuracy but significantly increases computational cost.
C2S should therefore be viewed not as a replacement for established bioinformatics pipelines, but as an interface layer between omics data and generative AI systems. Its strength lies in enabling language-model reasoning over biological data – while classical statistical and systems biology methods remain essential for quantitative rigor.
For biotech teams exploring production-grade integration of LLMs into drug discovery or multi-omics workflows, the key challenge is system design: data preprocessing, validation, evaluation metrics, and infrastructure alignment.
This is where structured implementation matters.

that balance biological fidelity, computational efficiency, and deployment constraints
Turn experimental frameworks into scalable research infrastructure