
There is a vast amount of natural proteins, but potentially, there are many more that can be created to address current challenges in biotechnology. The aim of protein engineering is to concept and produce proteins with defined properties and functions.
Traditionally, protein engineering has relied on two main methods:
- Directed evolution: creating a library with randomly mutated proteins and selecting those with desired traits. This approach is labor-intensive and time-consuming;
- Rational design: making modifications guided by structural and functional data. This is often limited by the quality and availability of structural information.
Artificial Intelligence is making this process significantly more efficient by shifting the paradigm from trial-and-error to a predictive, data-driven strategy.
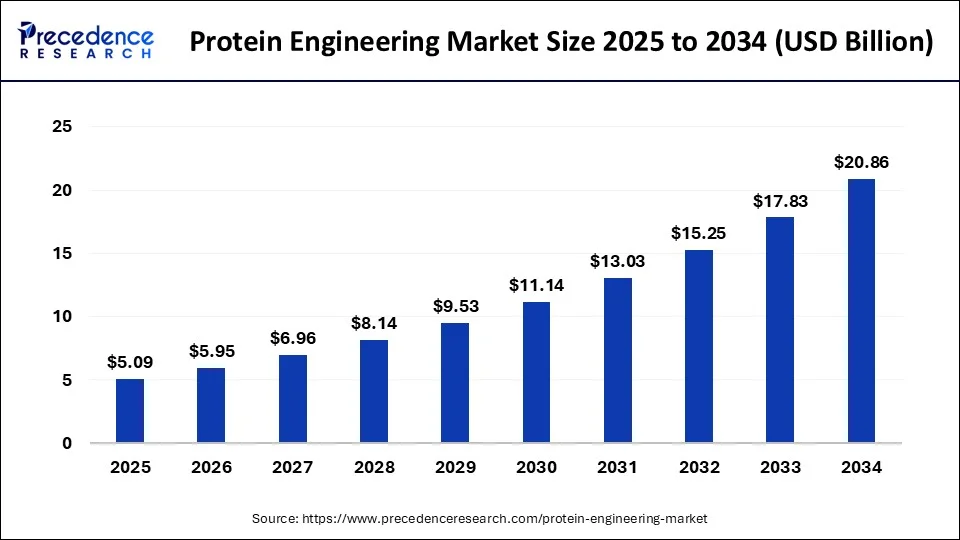
In 2024, this industry had a market size valued at USD 4.35 billion and is predicted to increase from USD 5.09 billion in 2025 to approximately USD 20.86 billion by 2034 (CAGR of 16.97%) (Precedence Research).
Unified scientific platform for multimodal protein data, predictive modeling, and generative therapeutic design.
Discover the CaseWhat Is Protein Engineering with AI?
Artificial intelligence can be useful in both protein engineering strategies:
- In directed evolution: propose mutations, predict function from sequence, substantially reducing experimental cycles;
- In rational design: without a pre-existing template or natural protein as a reference, using only biophysical and biochemical principles, can predict structure from sequence at near-experimental accuracy and enables de novo protein design (Koh et al., 2025).
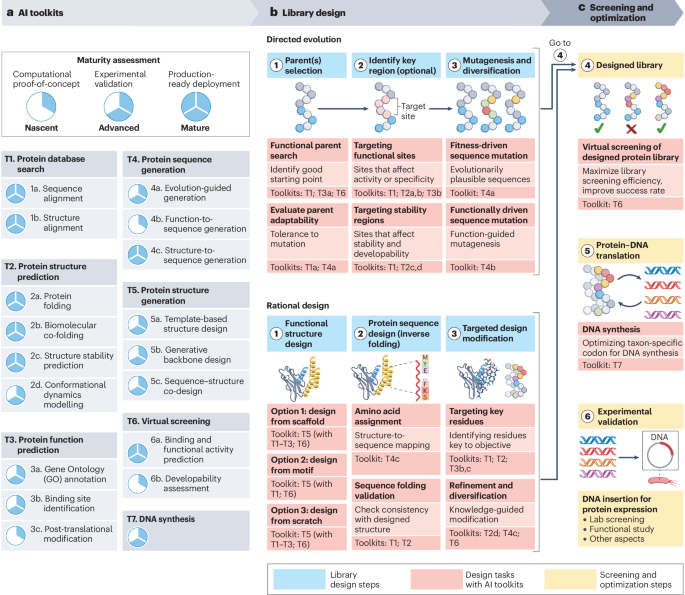
Let’s look closer at how AI tools can overcome key limitations and achieve better results.
Unprecedented Accuracy in Structure Prediction:
AI models extract coevolutionary patterns from homologous sequences, which leads to improved residue–residue contact maps and backbone geometry. By iteratively refining structural hypotheses and combining MSAs with 3D coordinate data, these tools achieve atomic-level predictions of experimental quality.
Newer Protein Language Models (PLMs) remove the requirement for Multiple Sequence Alignments (MSAs), allowing for faster, single-sequence structure inference. This is particularly valuable for orphan or rapidly evolving genes. Furthermore, by integrating neural network outputs with physics-based simulations, AI can better handle multi-domain proteins and complex topologies.
Expanding Protein Diversity:
AI helps overcome fixed-topology limitations, enabling the identification of novel fold architectures – including non-natural ones – and functional innovations beyond known PDB entries. This allows researchers to design entirely new protein scaffolds for specific families, avoiding the need for brute-force library screening. Crucially, AI keeps the core protein fold stable while optimizing loop regions, generating functionally meaningful diversity without random destabilization (Zhang et al., 2025).
Accelerating design cycles:
The process is driven by iterative loops where generative models propose novel sequences, and predictive tools evaluate their foldability and binding precision. GPU-accelerated inference allows for fast, cost-effective predictions at scale. These data-driven insights prioritize candidates with the highest probability of success by scoring catalytic efficiency, stability, solubility, and immune response properties, ultimately reducing the number of required physical assays.
AI-Powered Protein Engineering in Synthetic Biology:
AI has already demonstrated remarkable value in synthetic biology and biotechnology:
- Enhancement of non-natural catalytic reactions: high efficiency and stereoselectivity, stability in concentrated organic solvent conditions (up to 70% ethanol), and thermal resistance > 90 °C;
- Target and epitope specification: compact proteins and peptides with high affinity, stability, and efficacy (e.g., engineered miniproteins that neutralize snake venom toxins with 100% survival in affected mice);
- De novo design of enzymes: Kemp eliminase (top candidate is 60x better than initial design) and high success rates in generating candidates with desired activity (serine hydrolase: 20%, carbonic anhydrase: 35%);
- Better understanding of complex cell processes: engineered intracellular Ras–GTP activity sensors and proximity-labeling modules enabled analysis of resistance mechanisms to Ras-G12C inhibitors (Zhang et al., 2025).
AlphaFold3 Overview
Millions of researchers globally have used AlphaFold 2, and its scientific impact has been recognized through many prizes.
In 2024, Google DeepMind and Isomorphic Labs released an improved version – AlphaFold 3. They also launched AlphaFold Server to enable open access to AlphaFold, including a free database of 200 million protein structures (Google).
Main Advancements:
- High Accuracy: AF3 achieves a Global Distance Test (GDT) score of up to 90.1;
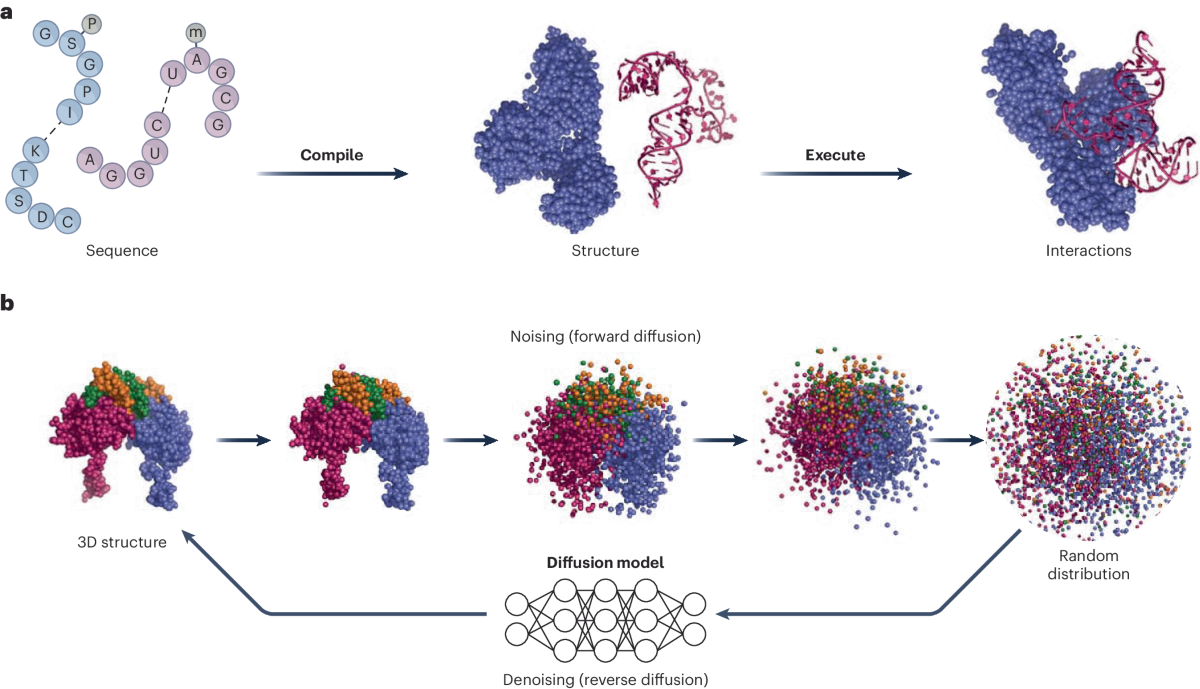
- Diffusion-Based Architecture: it predicts raw atomic coordinates by denoising random noise, capturing detailed structural features both locally and globally;
- Superior Precision: it offers 50% more precision compared to leading traditional methods on the PoseBusters benchmark.
Outperforms:
- AF2, Rosetta, I-TASSER, and Phyre2 across key parameters (RMSD, TM-score, pLDDT confidence, and computational time);
- Physics-based tools in predicting biomolecular structures (even without template structures);
- Traditional docking techniques in predicting protein–ligand interactions;
- Nucleic-acid-specific predictors in protein–nucleic acid interaction accuracy.
Key Capabilities:
- Chemical Modifications: Forecasting chemical modifications allows for a deeper understanding of cellular processes and disease connections;
- Speed: By using pair-weighted averaging and only 4 blocks of MSA, it significantly saves computational time;
- Flexibility: Doesn’t require excessive specialization for different molecule types;
- Precision: Approximates complex molecular interactions with angstrom accuracy;
- Functional Insights: Predictions correlate strongly with experimental data on protein stability and ligand binding affinities (r = 0.89);
- De Novo Design: When combined with GANs, it successfully generates artificial enzymes with desired catalytic activity.
Remaining Challenges
Despite its power, AF3 struggles with predicting dynamic protein behaviors and disordered regions. It may produce inaccurate structures for orphan proteins or those undergoing significant conformational changes upon ligand binding. Additionally, it sometimes generates overlapping atoms in large protein-nucleic complexes and is limited to predicting a single structure for a given sequence, missing alternative conformations (Abramson et al, 2024).

Get a personalized consultation with Ivan Izonin, PhD, Scientific Advisor in Artificial Intelligence at Blackthorn AI, to explore how advanced AI methods and next-generation models can accelerate your protein discovery, design, and biomedical research workflows.
Book a ConsultationBoltz 2 Overview
Accurately modeling biomolecular interactions is a critical property, but it hasn’t been enabled properly with any tools.
The Boltz team presented Boltz-2, which exhibits strong performance for both structure and affinity prediction. It’s freely available with open access to model weights, inference pipeline, and training code.
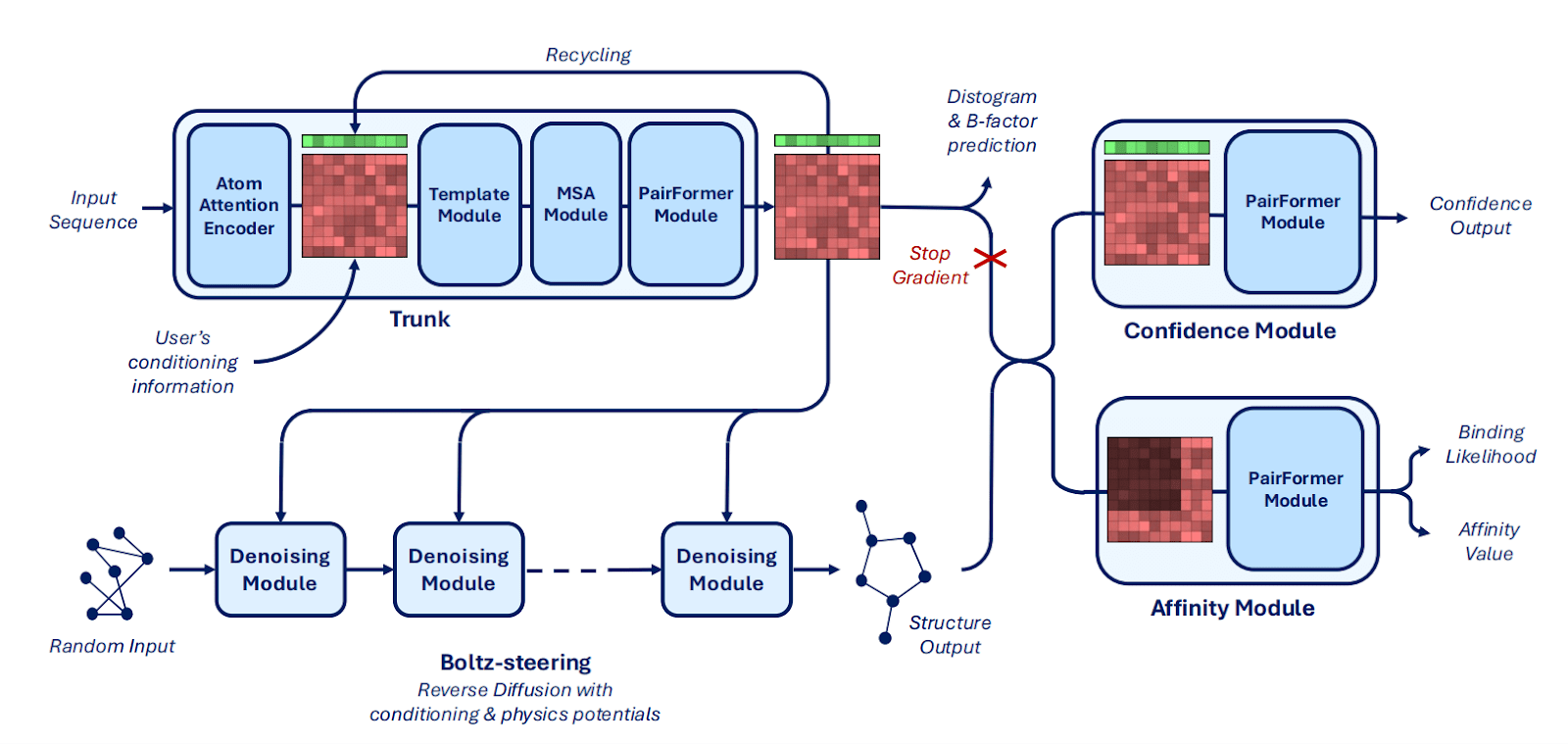
Main innovations in architecture:
Mixed-precision (bfloat16) and the trifast-4 kernel for triangle attention reduce runtime and memory use, enabling training with crop sizes up to 768 tokens.
Boltz-2x has Boltz-steering – an inference-time method that applies physics-based potentials, improves physical plausibility (overcomes steric clashes and incorrect stereochemistry).
Broader users’ controllability is achieved by integrating:
- Structure prediction method conditioning;
- Template conditioning and steering (integrates related complex structures or multimeric templates without retraining);
- Contact and pocket conditioning (allows using specific distance or pocket constraints);
- Specialized PairFormer refinement of protein–ligand contacts with dual-head prediction (one for binding likelihood and the other for continuous affinity) trained on heterogeneous affinity labels (Passaro et al., 2025).
Performance:
Boltz-2 outperforms methods like Haiping, GAT, and VincDeep in binding affinity prediction across 140 complexes. In hit discovery, it achieves double the average precision of standard ML and docking baselines.
Crucially, Boltz-2 is the first AI model to approach the performance of Free Energy Perturbation (FEP) methods, achieving a Pearson correlation of 0.62 (comparable to OpenFE) while being 1,000x more computationally efficient. It uses data curation and representation learning to overcome the performance/compute time trade-off.
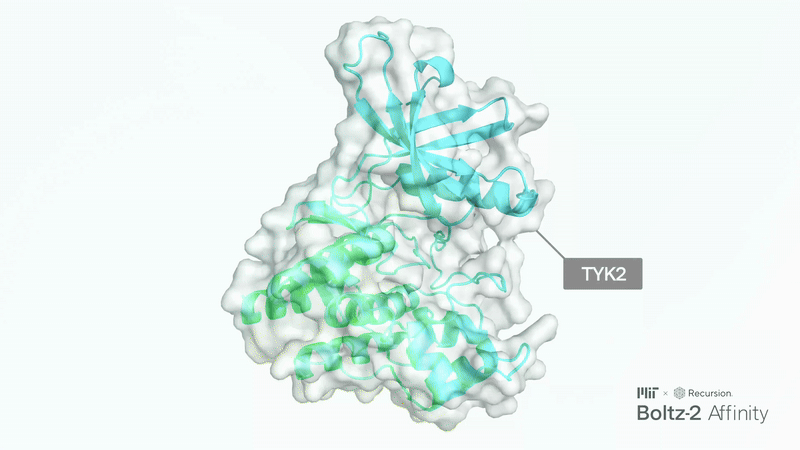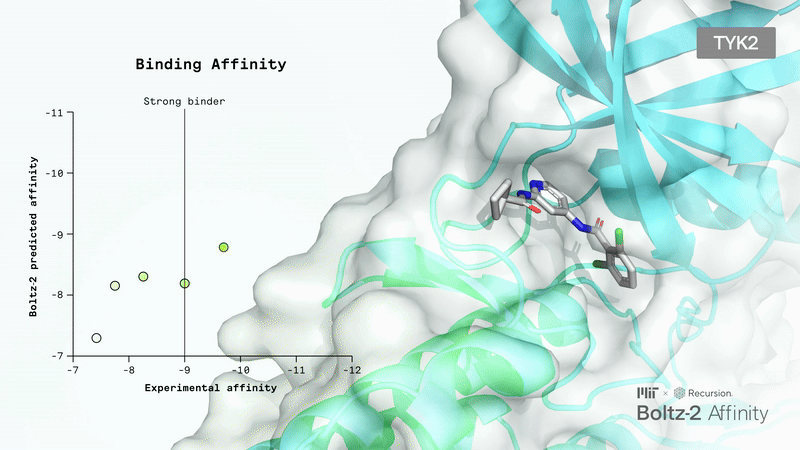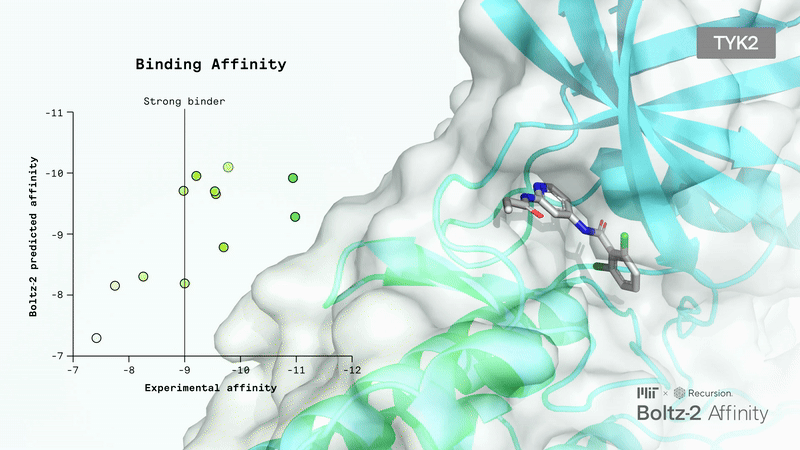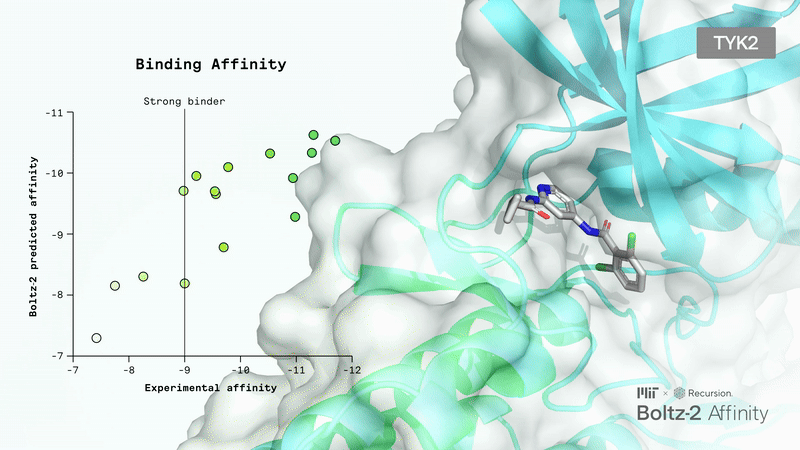
Limitations:
- Inefficient molecular dynamics (small dataset, minor architecture tweaks, limited multi-conformation handling);
- Trained on similar data as predecessors;
- Struggles with large complexes, cofactors (ions, water, or multimeric partners); may misplace parts of the ligand or generate chemically unrealistic conformations (requires additional help like templates or refinement steps);
- A limited affinity crop may truncate long-range interactions or miss relevant pockets;
- As a comparably new tool, performance has variability across assays, and robustness needs further testing (Passaro et al., 2025).
OpenFold3
OpenFold3 (OF3) is an open-source reimplementation of AlphaFold 3, designed to be permissive and accessible for industrial use. While currently in preview, the model aims to match AF3’s capabilities while offering a fully trainable, commercially usable framework.
Current Status and Federated Learning
While the OF3 preview does not yet match AF3 performance across all modalities, the team is actively improving it by training on newer PDB data, improving performance on weaker modalities (lDDT < 0.8), and speeding up inference.
A significant development is the Federated OpenFold3 Initiative. Since only about 2% of public protein structures are paired with drugs, five pharmaceutical companies will separately train OF3 on their proprietary libraries (approx. 4,000–8,000 protein-drug pairs each). Apheris will then aggregate these learnings. This collaborative approach aims to bridge the data gap in drug discovery (Saey, 2025).
OpenFold3 vs Boltz 2 vs AlphaFold3: Head-to-Head Comparison
| AlphaFold3 | Boltz-2 | OpenFold3 | |
| Prediction capabilities | – Structure of proteins, protein complexes (with another protein, nucleic acid, ligand/ion). – Post-translational modification. – High accuracy (especially for large complexes and multimeric assemblies). | – Structure of proteins, protein complexes (with protein, nucleic acid, ligand). – Binding affinity for protein-ligand interaction. – Approaches FEP (even 1000x faster). – High physical validity. | The same as AF3 |
| Training dataset | The exact dataset is unavailable. Reported sources: PDB (all structures up to 2021), Rfam (RNA), JASPAR/SELEX (protein–DNA), VDJdb (TCR–pMHC), IEDB (MHC–peptide epitopes), AlphaFold DB (a monomer distillation of about 5M proteins) | Experimental: All PDB entries released before 2023 (without complexes over 7MB or with more than 5000 residues). Binding affinity: MISATO (11,235 systems), ATLAS (1,284 proteins – 100 frames each), mdCATH (5,270 systems). Distillation data: RNA, protein-DNA/ligand, TCR-pMHC (explicit amount isn’t provided). NA-ligand (filtered 2500 examples). MHC (up to 100 sequences per allele for class I and up to 200 per allele pair for class II). AlphaFold DB (the same as AF3). | 300k experimental molecular structures > 40M synthetic structures∼ 20k protein-drug pairs will be added |
| Memory efficiency (maximum per GPU) | >5000 residues | ∼2400 residues | Not described yet |
| Framework | JAX: – Sophisticated automatic differentiation capabilities. – Beneficial for large-scale tensor operations. – Difficult for inexperienced users, less flexible. | PyTorch: – High accessibility, community support, and pre-built modules (can accelerate research). – Easier to debug and modify models. – Scalability across multiple GPUs. – Integration into existing workflows, creation of complex multi-component systems. | |
| Licensing | Creative Commons license – Prohibited commercial use without a license. – Some functionalities (ligand binding, certain kinds of modifications) aren’t fully available in the public version. | Permissive license – Allowed modification of the model. – Open commercial/academic usage and all functions are fully available. | |
| Weights and the full training pipeline/data | Under restricted access | Available | Will be available |
Comparison of AF3, Boltz2, and OpenFold (Google DeepMind (GitHub); OpenFold Consortium: OpenFold; Falk Hoffmann: Boltz-2 revolutionises drug discovery; Genophore : OpenFold vs AlphaFold2; NVIDIA: OpenFold/OpenFold2; Abramson et al., 2024; Passaro et al., 2025; The OpenFold3 Team; Naddaf, 2025; Saey, 2025).
Models experimental comparison
The evaluation will be of the most relevant and available models for multimeric protein structure prediction – Boltz-2, OF3, and AlphaFold Multimer (AFM).
To compare them, 10 structures (unseen by these models in the training sets) were used. Predictions were performed for oligomers, and then evaluated by the accuracy ofglobal structure (ipTM, TM), interface geometry (DockQ, Inter-chain iDDT, iRMSD), and inter-chain contacts (Jaccard index, Precision, Recall).
Metrics that were used:
- DockQ – evaluation of overall docking accuracy (< 0.23 – incorrect; 0.23 – 0.49 – acceptable; 0.49 – 0.8 – medium; ≥ 0.8 high quality) (Basu et al., 2016);
- iRMSD – interface atom deviation (> 4 Å low quality; 2–4 Å – acceptable; <2 Å – high quality) (Armougom et al., 2006);
- ipTM – predicted measure of interface similarity (< 0.6 likely to fail; 0.6 – 0.8 could be correct or wrong; > 0.8 confidence in high-quality prediction) (EMBL-EBI);
- TM-score – global structure similarity (< 0.2 random proteins; 0.2-0.5 likely not in the same fold; > 0.5 assume roughly the same fold) (Zhang et al., 2005);
- Inter-chain iDDT – per-residue inter-chain local distance difference (< 0.25 – incorrect interface; 0.25 – 0.5 – low quality; > 0.5 roughly correct) (Mariani et al., 2013);
- Jaccard index – overlap between predicted and native inter-chain contacts (0 – no shared contacts; 1 – perfect contact overlap);
- Precision – the proportion of predicted contacts that are correct (0 – 1 – higher value means fewer false positive contacts);
- Recall – the proportion of true native contacts that were recovered (0 – 1 – higher value means more true contacts captured).
AlphaFold Multimer
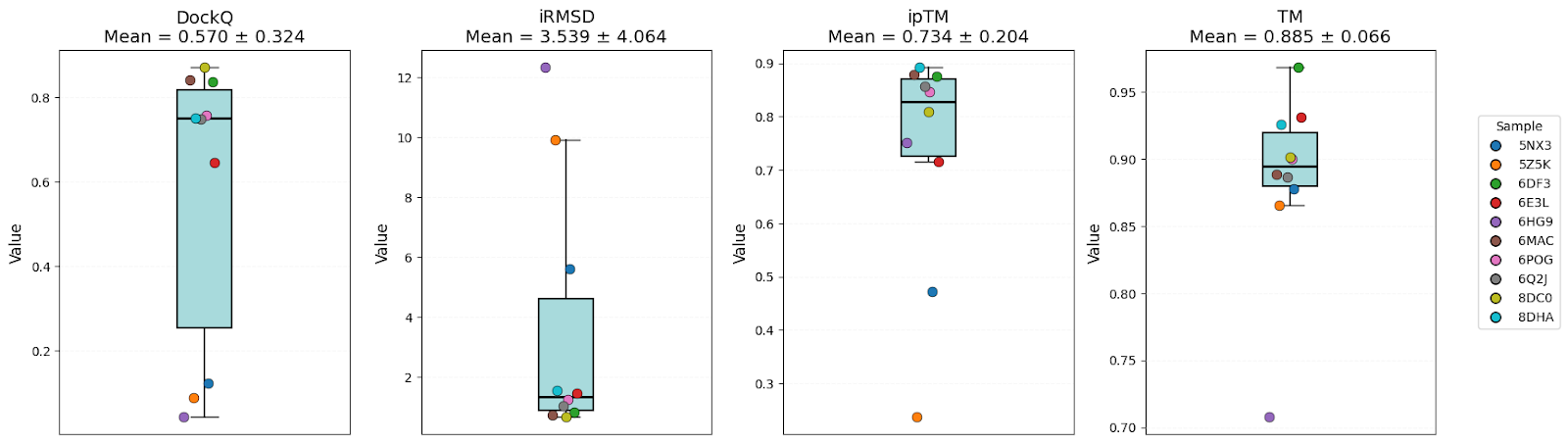
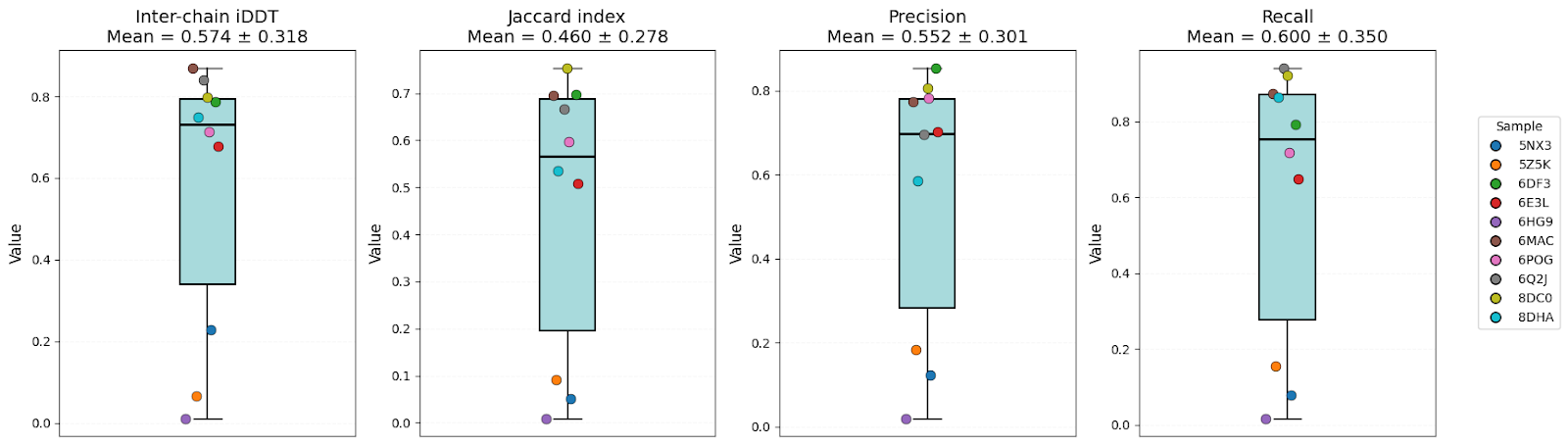
Boltz-2
OpenFold3
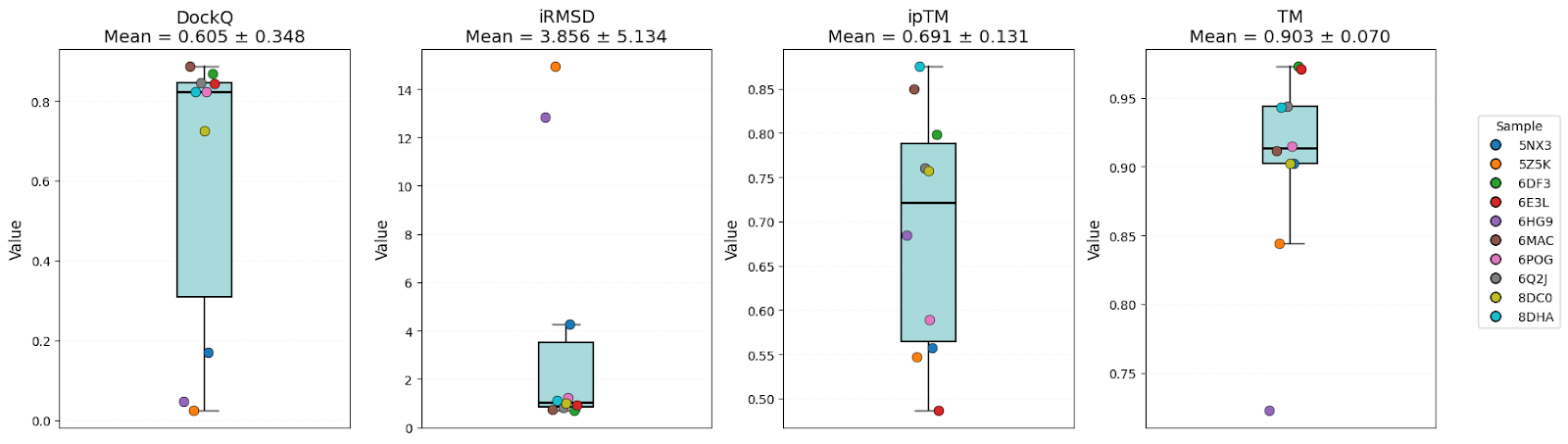
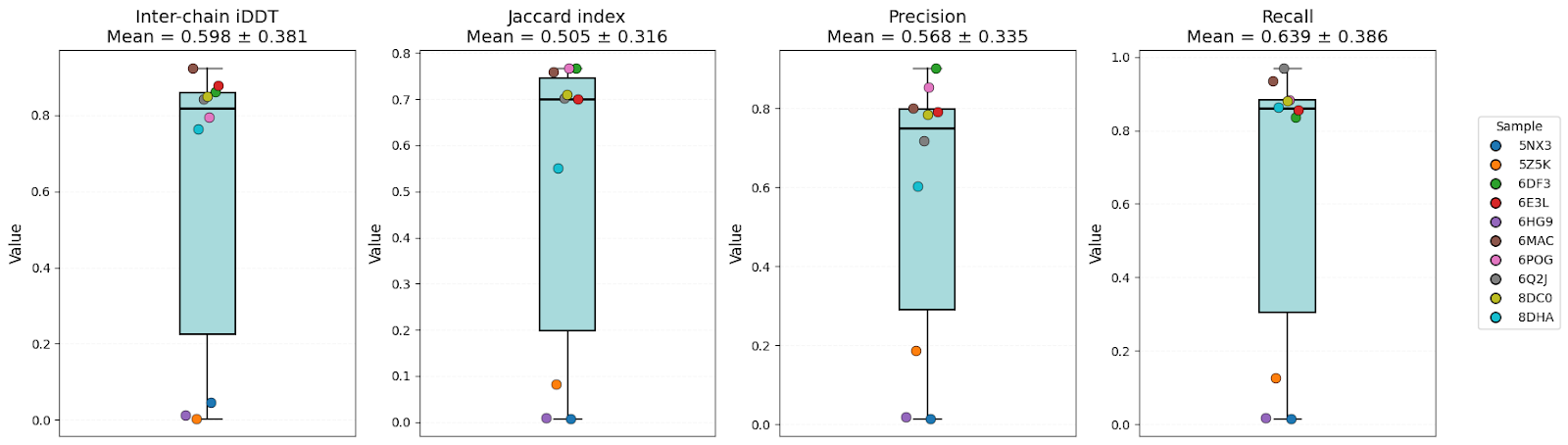
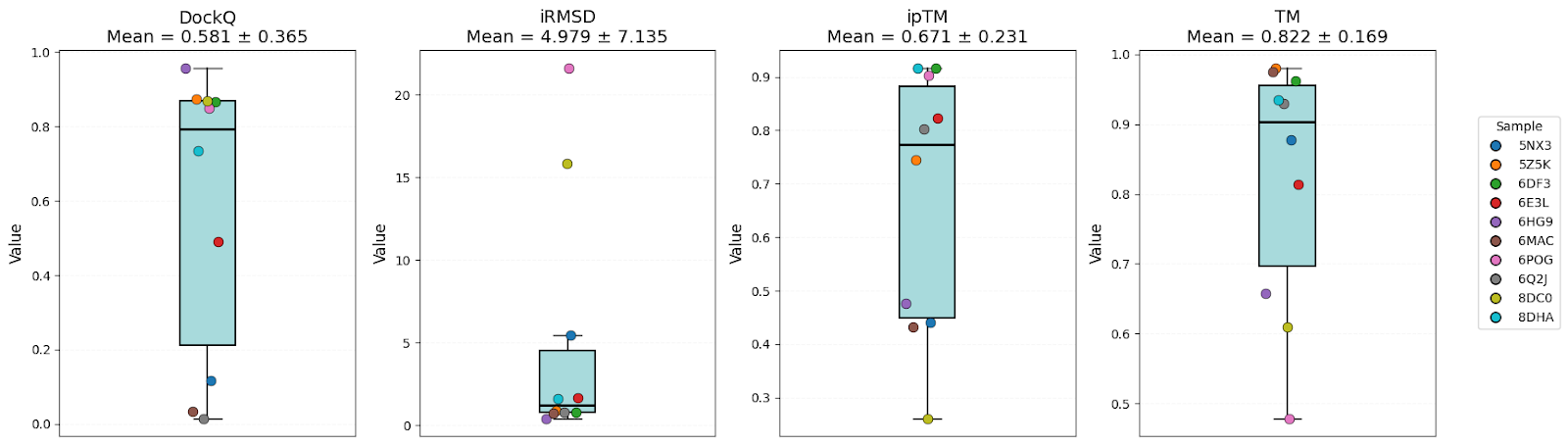
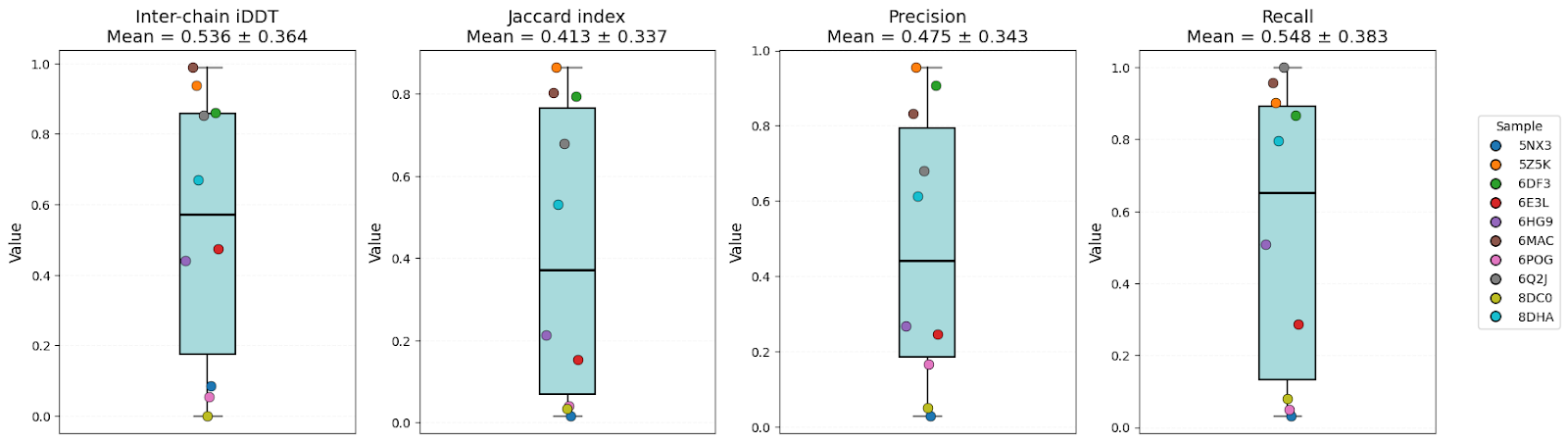
It’s clear that all models have high-level performance. However, in general, scores of AFM and OF3 were better and less variable, indicating consistency. Boltz-2 variability might be explained by the structure sensitivity reported in the available evaluations of this model.
| Score | AFM | Boltz-2 | OF3 | |
| (mean ± SD)1 | Better model | |||
| DockQ | 0.57 ± 0.32 | 0.58 ± 0.37 | 0.61 ± 0.35 | Equal |
| iRMSD | 3.54 ± 4.06 | 4.98 ± 7.14 | 3.86 ± 5.13 | AFM |
| ipTM | 0.73 ± 0.20 | 0.67 ± 0.23 | 0.69 ± 0.13 | AFM |
| TM-score | 0.89 ± 0.07 | 0.82 ± 0.17 | 0.90 ± 0.07 | AFM/OF3 |
| Inter-chain iDDT | 0.57 ± 0.32 | 0.54 ± 0.36 | 0.60 ± 0.38 | OF3 |
| Jaccard index | 0.46 ± 0.28 | 0.41 ± 0.34 | 0.51 ± 0.32 | AFM/OF3 |
| Precision | 0.55 ± 0.30 | 0.48 ± 0.34 | 0.57 ± 0.34 | AFM/OF3 |
| Recall | 0.60 ± 0.35 | 0.55 ± 0.38 | 0.64 ± 0.39 | OF3 |
Models have shown high structural accuracy, while having problems with interface geometry and recovering native inter-chain contacts. They not only miss a large number of true contacts but also predictmany false positives (especially Boltz-2).
On the other hand, Boltz-2 has a significantly lower level of clashes (aligns with its authors’ claim that this model has better physical validity) – only 10% with 1 clash per prediction. Surprisingly, OF3`s predictions have none, while 50% ofAFM’s predictions had 1-13 clashes.
Proportion of high-scored predictions:
| Score (with a threshold of high accuracy) | Percentage of predictions | Better model | ||
| AFM | Boltz-2 | OF3 | ||
| DockQ (≥ 0.8) | 30% | 50% | 60% | OF3 |
| iRMSD (< 2A) | 70% | 70% | 70% | Equal |
| ipTM (≥ 0.8) | 60% | 50% | 20% | AFM |
| TM-score (≥ 0.85) | 90% | 60% | 80% | AFM |
| Inter-chain iDDT (≥ 0.7) | 60% | 40% | 70% | OF3 |
| Jaccard index (≥ 0.5) | 70% | 50% | 70% | AFM/OF3 |
| Precision (≥ 0.5) | 70% | 50% | 70% | AFM/OF3 |
| Recall (≥ 0.5) | 70% | 60% | 70% | AFM/OF3 |
Such a distribution additionally confirms that AFM and OF3 are less variable. AFM’s percentage of high DockQ might be surprising, but it’s important to take into account that its scores didn’t achieve that high level as Boltz-2, but the last model also has more samples with low DockQ scores. Thus, this score also follows a general pattern. The same situation with OF3`s ipTM scores.
To summarise, AFM and OF3 have more stable and better performance over all measured scores, but AFM is inferior to Boltz-2 and OF3 in handling steric clashes. Additionally, sometimes Boltz-2 has higher accuracy than compared models, but this is offset by a higher level of low-quality predictions.
Conclusion
AI has arrived as a timely solution for the pharmaceutical industry, addressing challenges of cost, efficiency, and low success rates in traditional protein engineering.
- AlphaFold 3 remains the benchmark for accuracy but is limited by restrictive licensing and “black box” constraints;
- Boltz-2 offers a powerful, open-source alternative that excels in binding affinity and physical plausibility;
- OpenFold3 promises to democratize AF3’s architecture for commercial use, though it is still refining its performance.
The future of protein engineering depends not only on improving individual models but on integrating their complementary strengths into accessible, versatile platforms.
