Chemical Sciences
Chemical reaction yield prediction
Business Goals
Develop an application for estimating the chemical yield of the target product for unexplored organic reactions.
Challenge
Typically, product yield estimations are empirical, i.e., based on synthetic chemists’ experience. Therefore, there is no reliable way to predict yields for new chemical reactions.
Results
- Our approach outperforms or equals all known state-of-the-art AI models predicting chemical reaction yields on public datasets:
- R^2 = 0.86, RMSE = 1.35 on Suzuki-Miyaura [https://www.science.org/doi/10.1126/science.aap9112]
- R^2 = 0.93 on Buchwald-Hartwig [https://www.science.org/doi/10.1126/science.aar5169]
Implementation Details
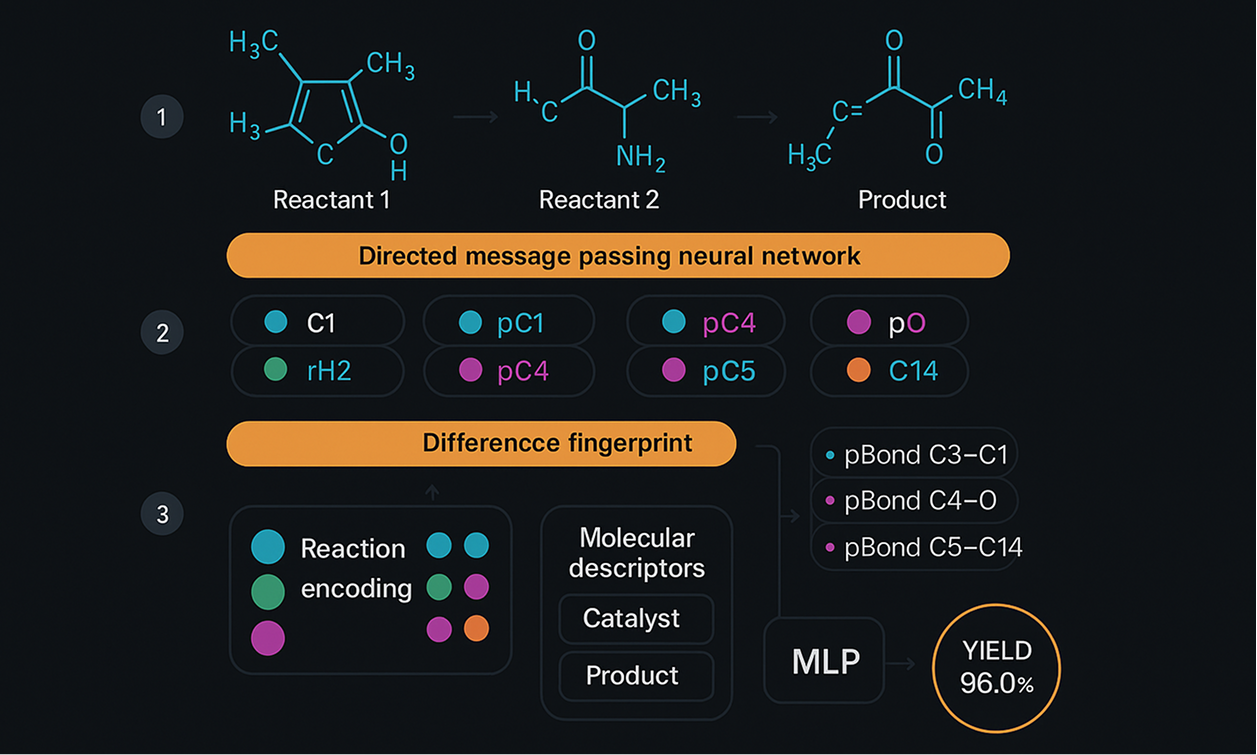
- A novel graph neural network architecture (RD-MPNN) was designed for organic reaction yield prediction. The network combines structural information, molecular-, and reaction-level descriptors. The network’s performance was compared with the performance of other machine learning models on the same data and targets: linear models, decision tree ensembles, fully-connected feed-forward, and transformer networks. The RD-MPNN outperformed all the listed approaches in single- and multi-reaction class settings.

Alex Gurbych
Chief Solutions Architect
Receive a professional and in-depth consultation from an experienced expert. Get tailored advice to address your specific needs and achieve your goals effectively.